Please note that the fine-tuning examples are meant to be generally applicable and the input datasets and labels will vary dependent on the downstream task. Example input files for a few of the downstream tasks demonstrated in the manuscript are located within the [example_input_files directory](https://huggingface.co/datasets/ctheodoris/Genecorpus-30M/tree/main/example_input_files) in the dataset repository, but these only represent a few example fine-tuning applications.
Please note that GPU resources are required for efficient usage of Geneformer. Additionally, we strongly recommend tuning hyperparameters for each downstream fine-tuning application as this can significantly boost predictive potential in the downstream task (e.g. max learning rate, learning schedule, number of layers to freeze, etc.).
## 模型下载
### geneformer模型下载
datasets: ctheodoris/Genecorpus-30M
license: apache-2.0
---
# Geneformer
Geneformer is a foundation transformer model pretrained on a large-scale corpus of ~30 million single cell transcriptomes to enable context-aware predictions in settings with limited data in network biology.
模型下载以及安装geneformls
- See [our manuscript](https://rdcu.be/ddrx0) for details.
- See [geneformer.readthedocs.io](https://geneformer.readthedocs.io) for documentation.
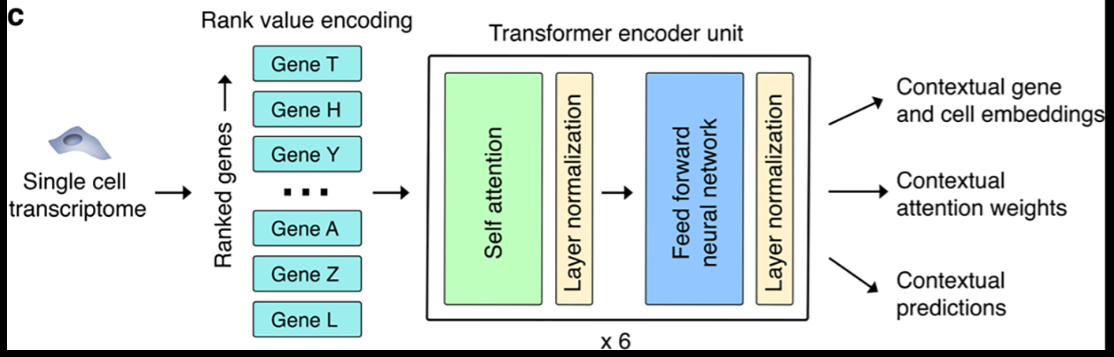
Geneformer is a foundation transformer model pretrained on [Genecorpus-30M](https://huggingface.co/datasets/ctheodoris/Genecorpus-30M), a pretraining corpus comprised of ~30 million single cell transcriptomes from a broad range of human tissues. We excluded cells with high mutational burdens (e.g. malignant cells and immortalized cell lines) that could lead to substantial network rewiring without companion genome sequencing to facilitate interpretation. Each single cell’s transcriptome is presented to the model as a rank value encoding where genes are ranked by their expression in that cell normalized by their expression across the entire Genecorpus-30M. The rank value encoding provides a nonparametric representation of that cell’s transcriptome and takes advantage of the many observations of each gene’s expression across Genecorpus-30M to prioritize genes that distinguish cell state. Specifically, this method will deprioritize ubiquitously highly-expressed housekeeping genes by normalizing them to a lower rank. Conversely, genes such as transcription factors that may be lowly expressed when they are expressed but highly distinguish cell state will move to a higher rank within the encoding. Furthermore, this rank-based approach may be more robust against technical artifacts that may systematically bias the absolute transcript counts value while the overall relative ranking of genes within each cell remains more stable.
The rank value encoding of each single cell’s transcriptome then proceeds through six transformer encoder units. Pretraining was accomplished using a masked learning objective where 15% of the genes within each transcriptome were masked and the model was trained to predict which gene should be within each masked position in that specific cell state using the context of the remaining unmasked genes. A major strength of this approach is that it is entirely self-supervised and can be accomplished on completely unlabeled data, which allows the inclusion of large amounts of training data without being restricted to samples with accompanying labels.
We detail applications and results in [our manuscript](https://rdcu.be/ddrx0).
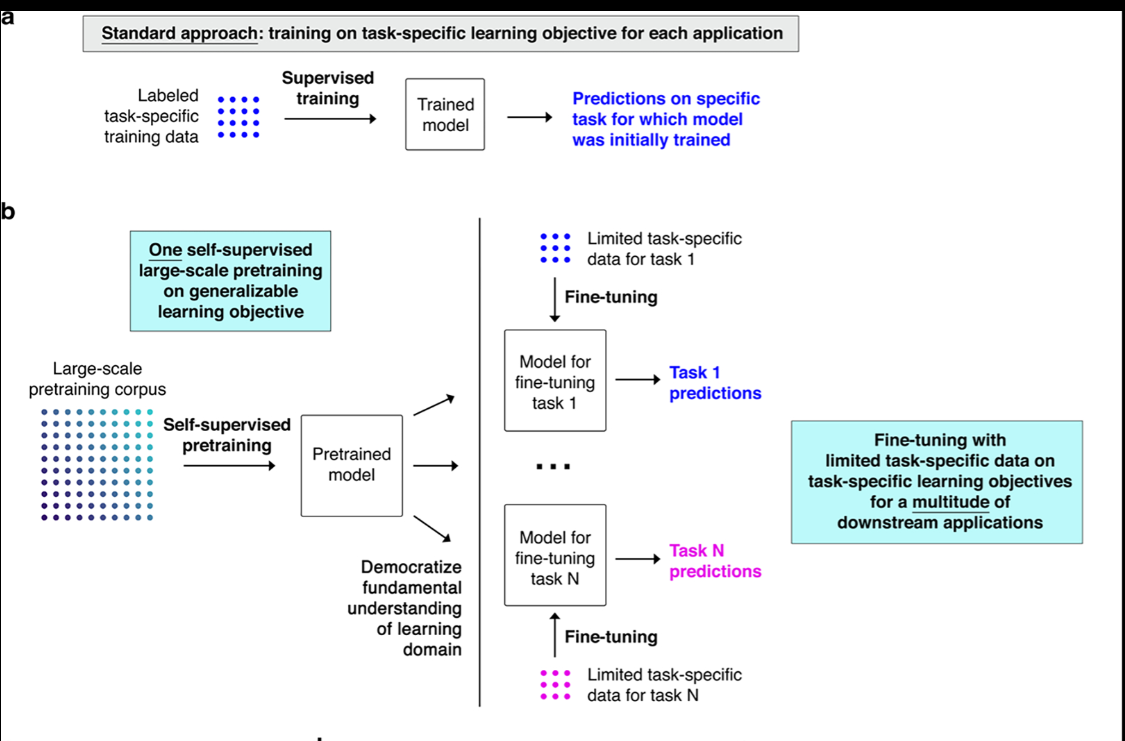
During pretraining, Geneformer gained a fundamental understanding of network dynamics, encoding network hierarchy in the model’s attention weights in a completely self-supervised manner. With both zero-shot learning and fine-tuning with limited task-specific data, Geneformer consistently boosted predictive accuracy in a diverse panel of downstream tasks relevant to chromatin and network dynamics. In silico perturbation with zero-shot learning identified a novel transcription factor in cardiomyocytes that we experimentally validated to be critical to their ability to generate contractile force. In silico treatment with limited patient data revealed candidate therapeutic targets for cardiomyopathy that we experimentally validated to significantly improve the ability of cardiomyocytes to generate contractile force in an iPSC model of the disease. Overall, Geneformer represents a foundational deep learning model pretrained on ~30 million human single cell transcriptomes to gain a fundamental understanding of gene network dynamics that can now be democratized to a vast array of downstream tasks to accelerate discovery of key network regulators and candidate therapeutic targets.
In [our manuscript](https://rdcu.be/ddrx0), we report results for the 6 layer Geneformer model pretrained on Genecorpus-30M. We additionally provide within this repository a 12 layer Geneformer model, scaled up with retained width:depth aspect ratio, also pretrained on Genecorpus-30M.
# 模型训练
Both the 6 and 12 layer Geneformer models were pretrained in June 2021.
单卡运行 gene classification
```
# Application
The pretrained Geneformer model can be used directly for zero-shot learning, for example for in silico perturbation analysis, or by fine-tuning towards the relevant downstream task, such as gene or cell state classification.
Example applications demonstrated in [our manuscript](https://rdcu.be/ddrx0) include:

{kind=link}
{kind=link}