"tests/vscode:/vscode.git/clone" did not exist on "fb02f39ad8736da962951ecf54658dd1881b902f"

Merge pull request #439 from aqlaboratory/setup-improvements
Adds Documentation and minor quality of life fixes
Showing
.readthedocs.yaml
0 → 100644
This diff is collapsed.
docs/Makefile
0 → 100644
docs/environment.yml
0 → 100644
docs/imgs/of_banner.png
0 → 100755
{kind=link}
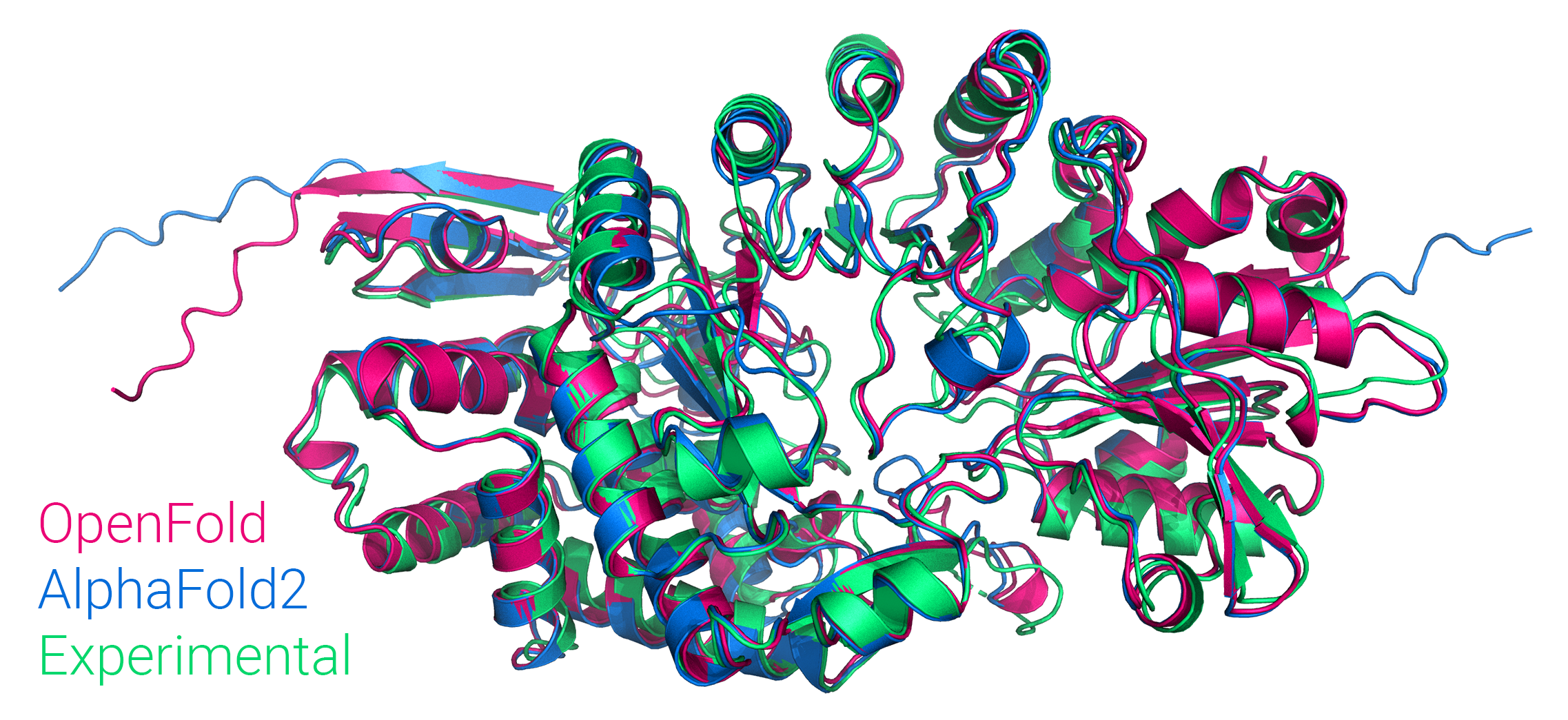
1.84 MB
docs/source/Aux_seq_files.md
0 → 100644
docs/source/FAQ.md
0 → 100644
docs/source/Inference.md
0 → 100644
docs/source/conf.py
0 → 100644
docs/source/index.md
0 → 100644
docs/source/installation.md
0 → 100644
This diff is collapsed.