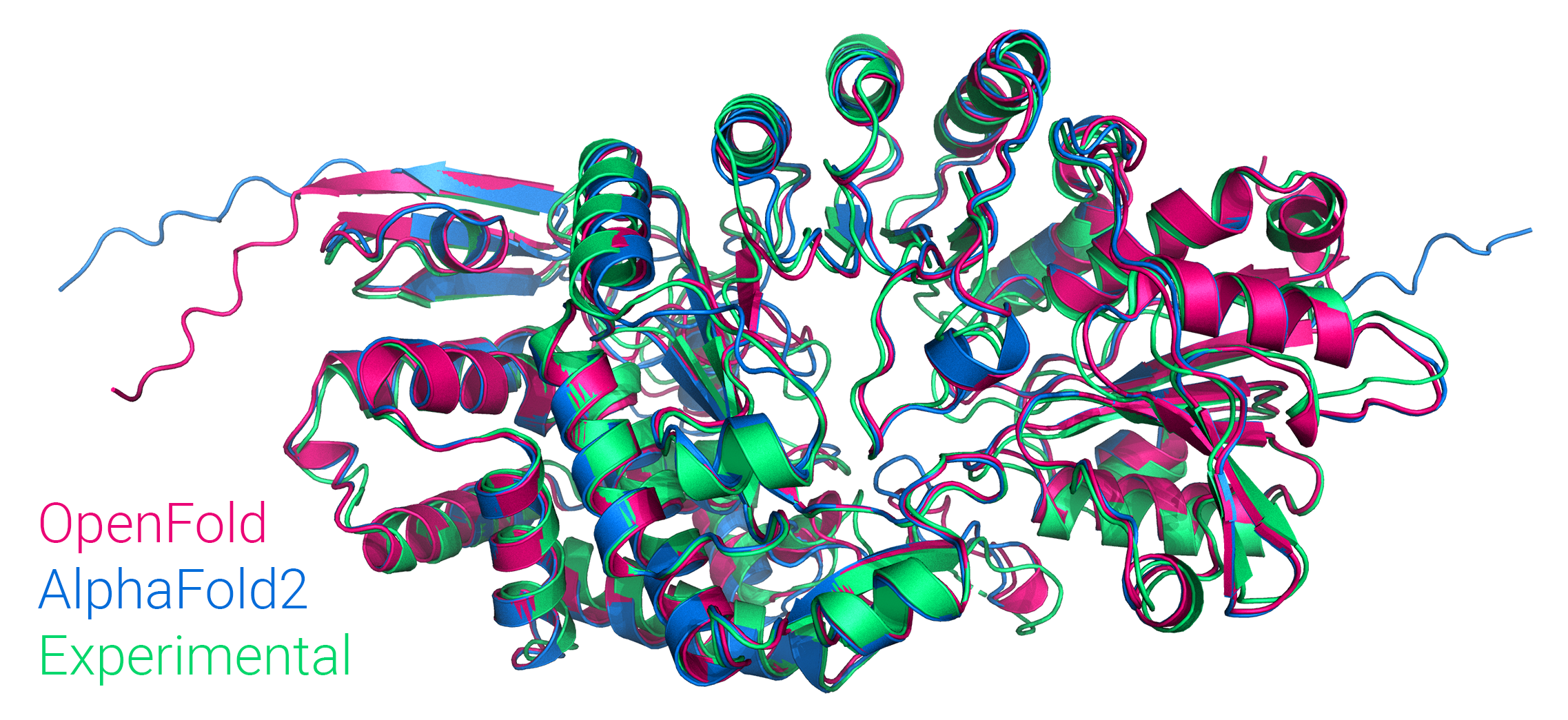
and we've trained it from scratch, matching the performance of the original.
We've publicly released model weights and our training data — some 400,000
MSAs and PDB70 template hit files — under a permissive license. Model weights
are available via scripts in this repository while the MSAs are hosted by the
[Registry of Open Data on AWS (RODA)](https://registry.opendata.aws/openfold).
Try out running inference for yourself with our [Colab notebook](https://colab.research.google.com/github/aqlaboratory/openfold/blob/main/notebooks/OpenFold.ipynb).
Additionally, OpenFold has the following advantages over the reference implementation:
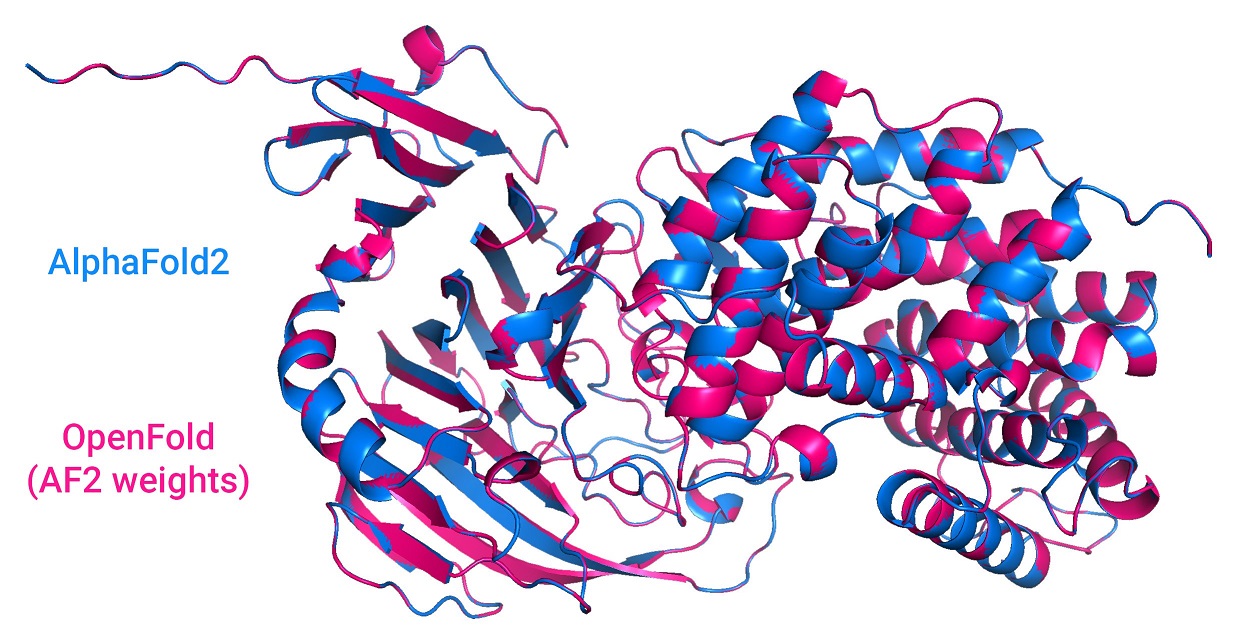
OpenFold also supports inference using AlphaFold's official parameters.
- Openfold is **trainable** in full precision or `bfloat16` half-precision, with or without [DeepSpeed](https://github.com/microsoft/deepspeed).
OpenFold has the following advantages over the reference implementation:
-**Faster inference** on GPU.
-**Faster inference** on GPU for chains with < 1500 residues.
-**Inference on extremely long chains**, made possible by our implementation of low-memory attention
-**Inference on extremely long chains**, made possible by our implementation of low-memory attention
([Rabe & Staats 2021](https://arxiv.org/pdf/2112.05682.pdf)). OpenFold can predict the structures of
sequences with more than 4000 residues on a single A100, and even longer ones with CPU offloading.
-**Custom CUDA attention kernels** modified from [FastFold](https://github.com/hpcaitech/FastFold)'s
-**Custom CUDA attention kernels** modified from [FastFold](https://github.com/hpcaitech/FastFold)'s
kernels support in-place attention during inference and training. They use
kernels support in-place attention during inference and training. They use
4x and 5x less GPU memory than equivalent FastFold and stock PyTorch
4x and 5x less GPU memory than equivalent FastFold and stock PyTorch
implementations, respectively.
implementations, respectively.
-**Efficient alignment scripts** using the original AlphaFold HHblits/JackHMMER pipeline or [ColabFold](https://github.com/sokrypton/ColabFold)'s, which uses the faster MMseqs2 instead. We've used them to generate millions of alignments that will be released alongside original OpenFold weights, trained from scratch using our code (more on that soon).
-**Efficient alignment scripts** using the original AlphaFold HHblits/JackHMMER pipeline or [ColabFold](https://github.com/sokrypton/ColabFold)'s, which uses the faster MMseqs2 instead. We've used them to generate millions of alignments.
## Installation (Linux)
## Installation (Linux)
...
@@ -70,7 +77,7 @@ To install the HH-suite to `/usr/bin`, run
...
@@ -70,7 +77,7 @@ To install the HH-suite to `/usr/bin`, run
## Usage
## Usage
To download DeepMind's pretrained parameters and common ground truth data, run:
To download the databases used to train OpenFold and AlphaFold run:
```bash
```bash
bash scripts/download_data.sh data/
bash scripts/download_data.sh data/
...
@@ -96,12 +103,13 @@ Make sure to run the latter command on the machine that will be used for MSA
...
@@ -96,12 +103,13 @@ Make sure to run the latter command on the machine that will be used for MSA
generation (the script estimates how the precomputed database index used by
generation (the script estimates how the precomputed database index used by
MMseqs2 should be split according to the memory available on the system).
MMseqs2 should be split according to the memory available on the system).
Alternatively, you can use raw MSAs from
Alternatively, you can use raw MSAs from our aforementioned MSA database or
[ProteinNet](https://github.com/aqlaboratory/proteinnet). After downloading
[ProteinNet](https://github.com/aqlaboratory/proteinnet). After downloading
the database, use `scripts/prep_proteinnet_msas.py` to convert the data into
the latter database, use `scripts/prep_proteinnet_msas.py` to convert the data
a format recognized by the OpenFold parser. The resulting directory becomes the
into a format recognized by the OpenFold parser. The resulting directory
`alignment_dir` used in subsequent steps. Use `scripts/unpack_proteinnet.py` to
becomes the `alignment_dir` used in subsequent steps. Use
extract `.core` files from ProteinNet text files.
`scripts/unpack_proteinnet.py` to extract `.core` files from ProteinNet text
files.
For both inference and training, the model's hyperparameters can be tuned from
For both inference and training, the model's hyperparameters can be tuned from
`openfold/config.py`. Of course, if you plan to perform inference using
`openfold/config.py`. Of course, if you plan to perform inference using

{kind=link}
{kind=link}