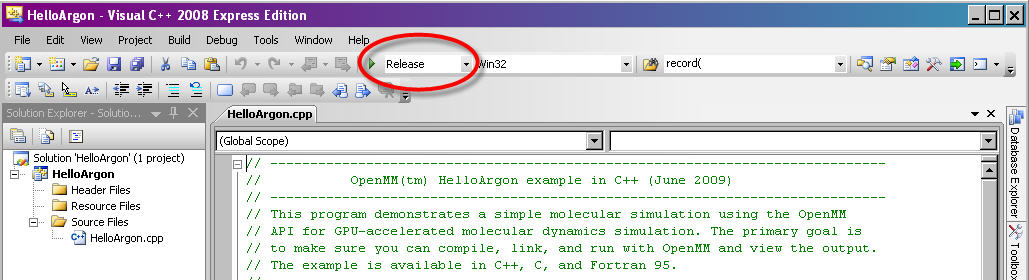
#. Wherever the C++ API uses references to return multiple values from a method, the Python API returns a tuple. For example, in C++ you would query a HarmonicBondForce for a bond’s parameters as follows:
title = {Computer Simulation of Biomolecular Systems},
editor = {Wilkinson, A. and Weiner, P. and van Gunsteren, Wilfred F.},
publisher = {Elsevier},
volume = {3},
pages = {83-96},
year = {1997},
type = {Book Section}
}
@article{Labute2008
author={Labute,Paul},
title = {The generalized Born/volume integral implicit solvent model: Estimation of the free energy of hydration using London dispersion instead of atomic surface area},
title = {A generalized reaction field method for molecular dynamics simulations},
journal = {Journal of Chemical Physics},
volume = {102},
number = {13},
pages = {5451-5459},
year = {1995},
type = {Journal Article}
}
@article{Toukmaji1996
author={Toukmaji,AbdulnourY.andBoardJr,JohnA.},
title = {Ewald summation techniques in perspective: a survey},
journal = {Computer Physics Communications},
volume = {95},
pages = {73-92},
year = {1996},
type = {Journal Article}
}
@article{Wang2000
author={Wang,J.andCieplak,P.andKollman,P.A.},
title = {How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules?},
where :math:`\alpha`, :math:`\beta`, and :math:`\gamma` are the GB\ :sup:`OBC`\ II parameters :math:`\alpha` = 1, :math:`\beta` = 0.8, and :math:`\gamma` =
4.85. :math:`\rho_i` is the adjusted atomic radius of particle *i*\ , which
is calculated from the atomic radius :math:`r_i` as :math:`\rho_i = r_i-0.009` nm.
:math:`\Psi_i` is calculated as an integral over the van der Waals

{kind=link}
{kind=link}
{kind=link}
{kind=link}