
Renamed docs to docs-source
Showing
{kind=link}

48 KB
docs-source/images/Argon.png
0 → 100644
{kind=link}

32.8 KB
{kind=link}
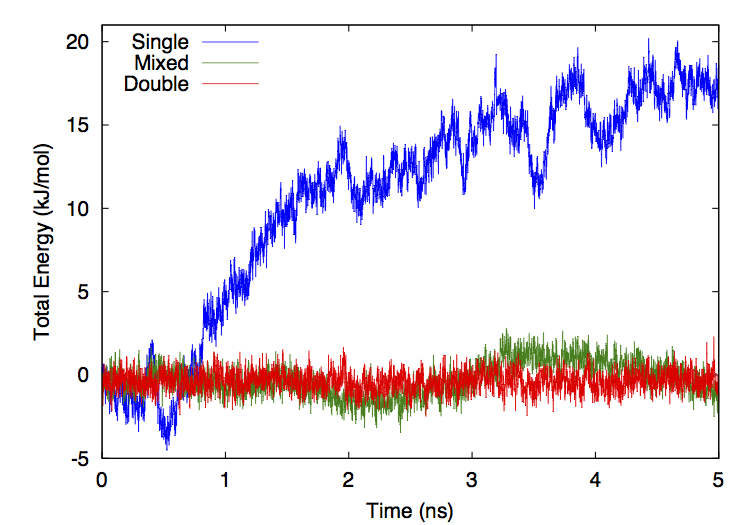
130 KB
{kind=link}
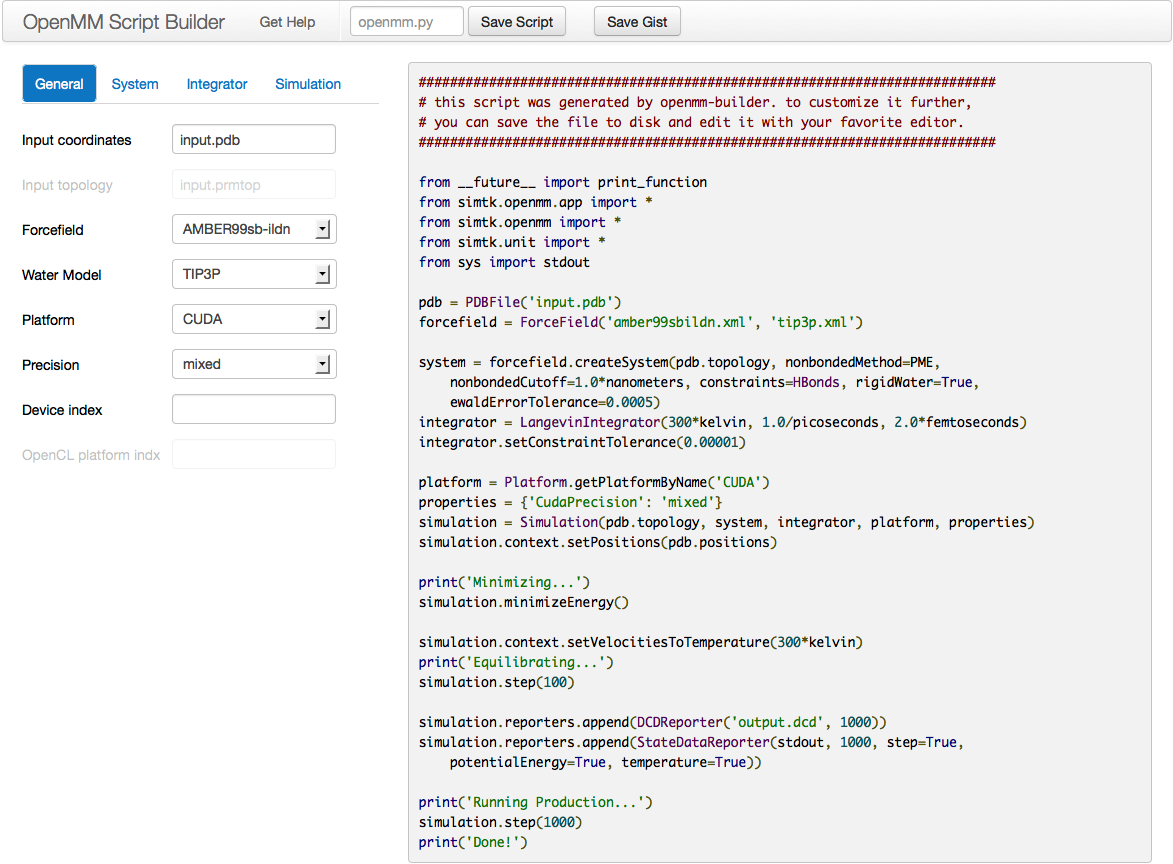
259 KB
{kind=link}
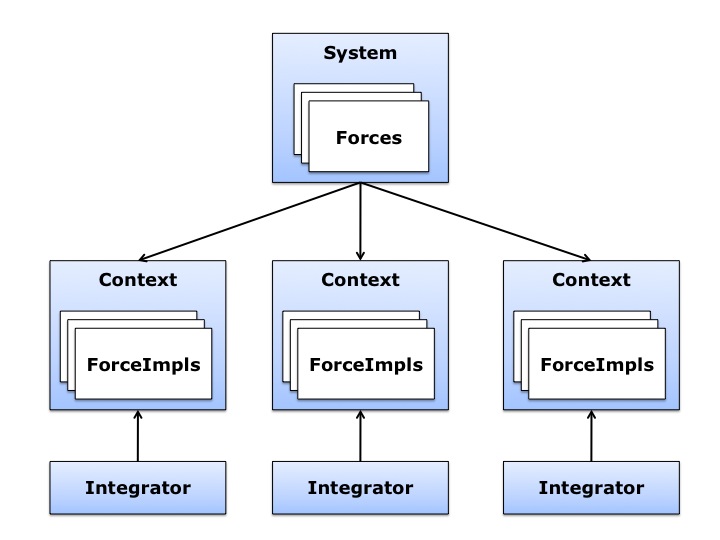
57.5 KB
{kind=link}
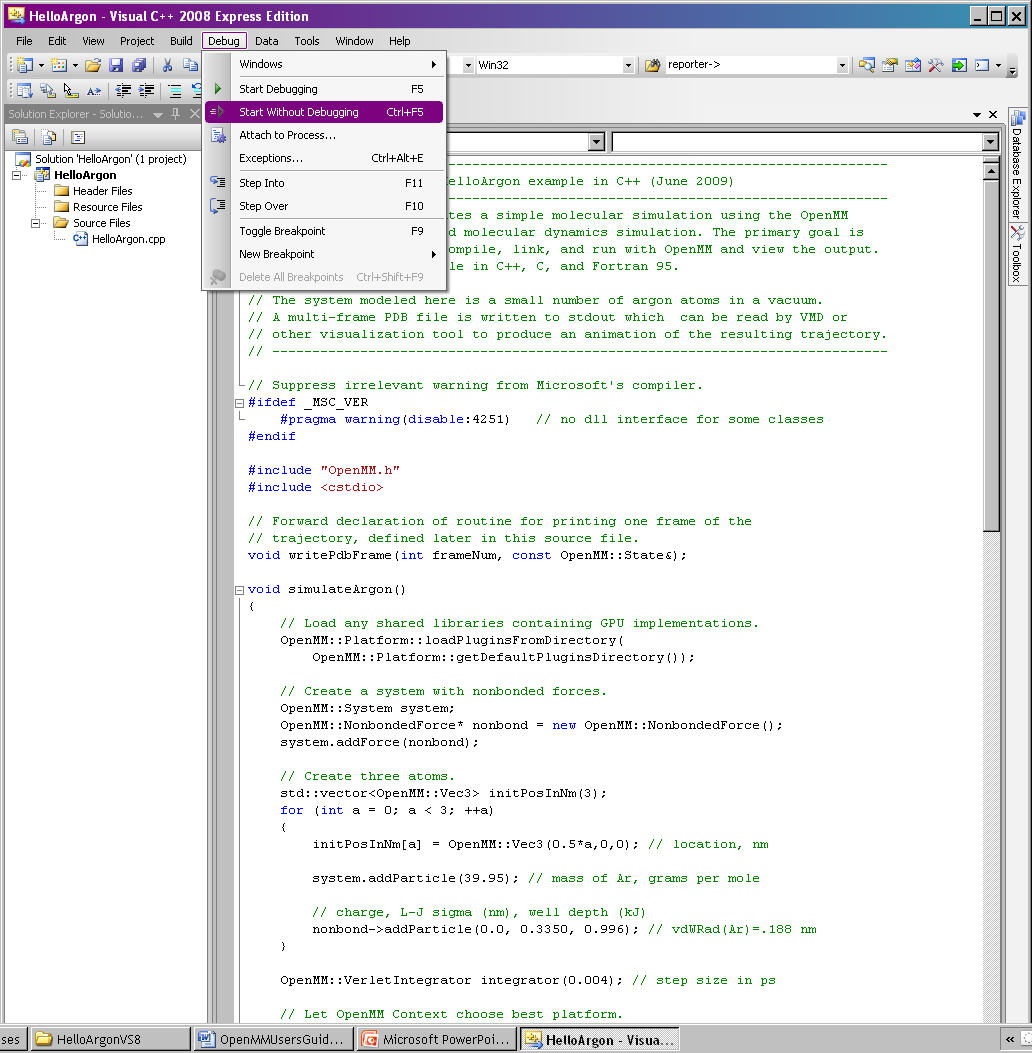
220 KB
{kind=link}
88 KB
docs-source/licenses/GPL.txt
0 → 100644
docs-source/sphinx/numsec.py
0 → 100644