
dtk24.04.1
parents
Showing
docs/alphafold2_1.png
0 → 100644
{kind=link}
140 KB
docs/casp15_predictions.zip
0 → 100644
File added
image.png
0 → 100644
{kind=link}
130 KB
imgs/casp14_predictions.gif
0 → 100644
{kind=link}
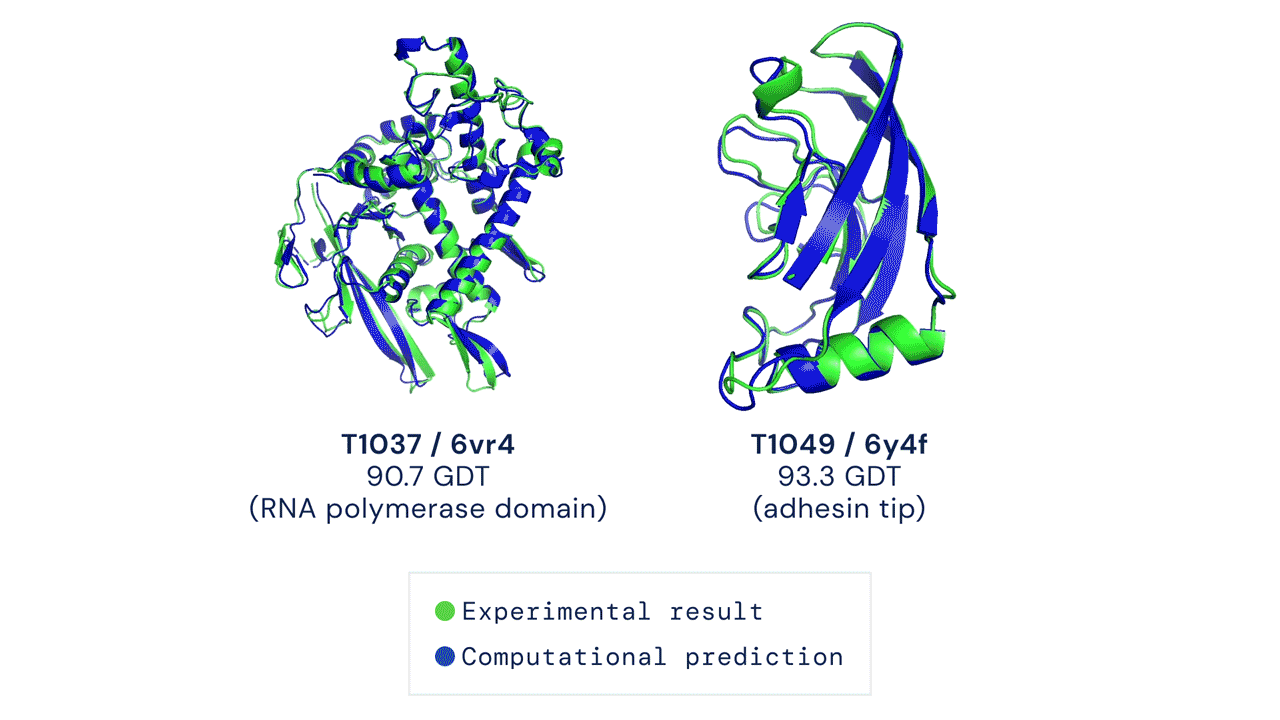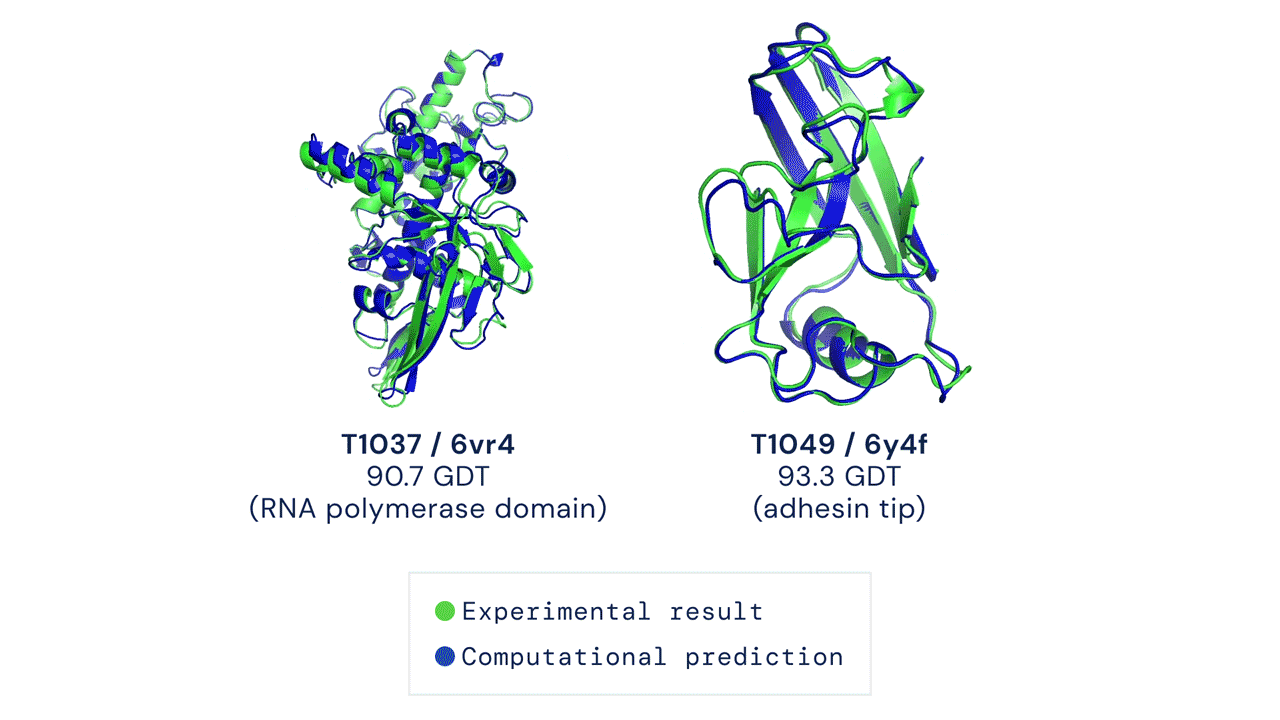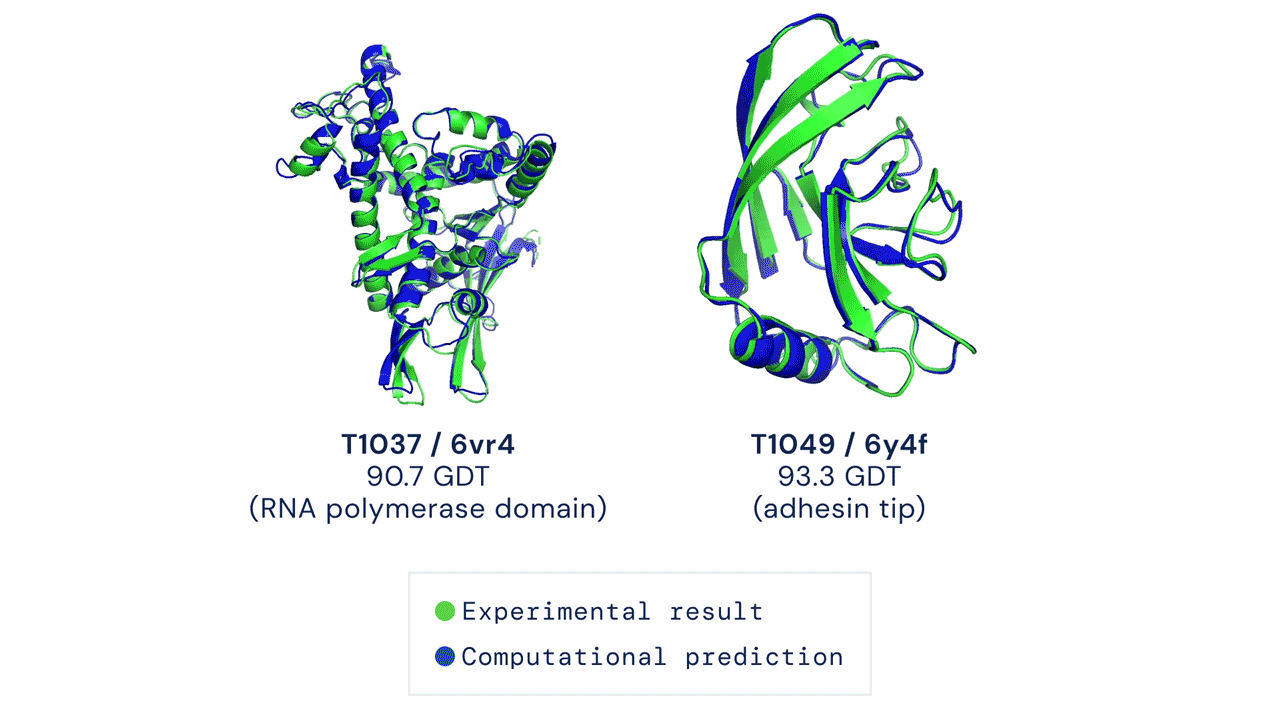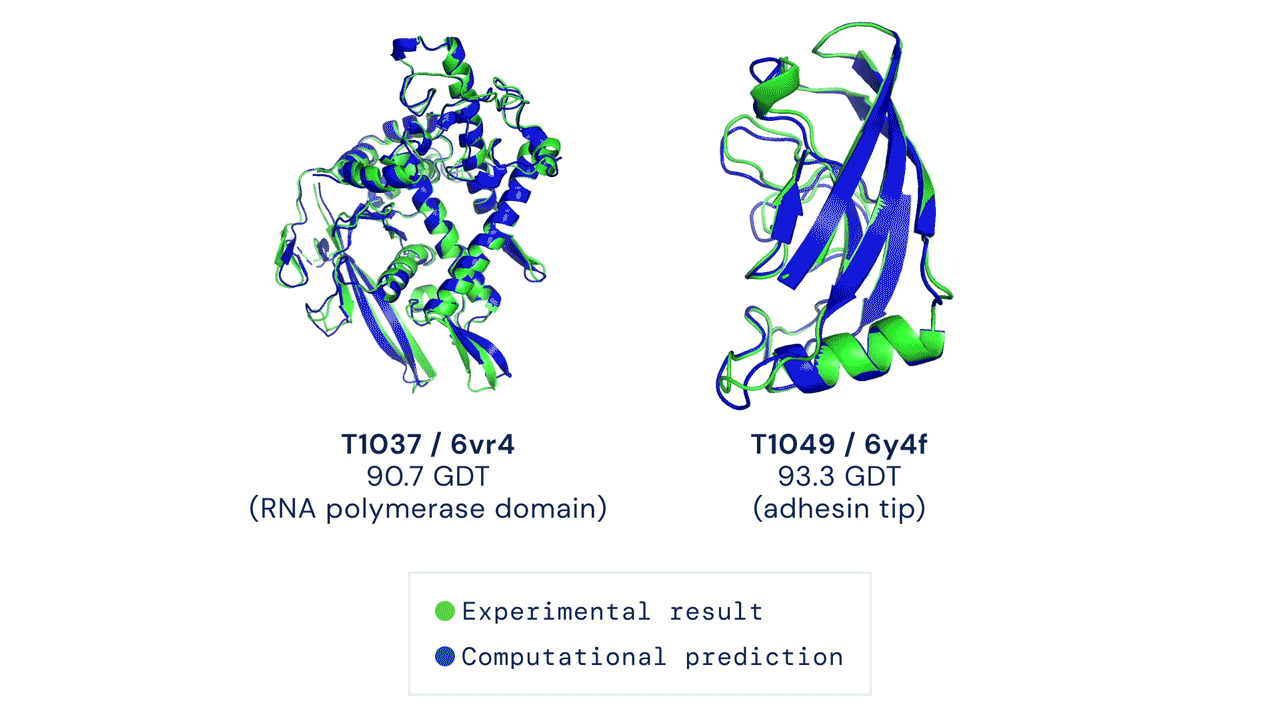
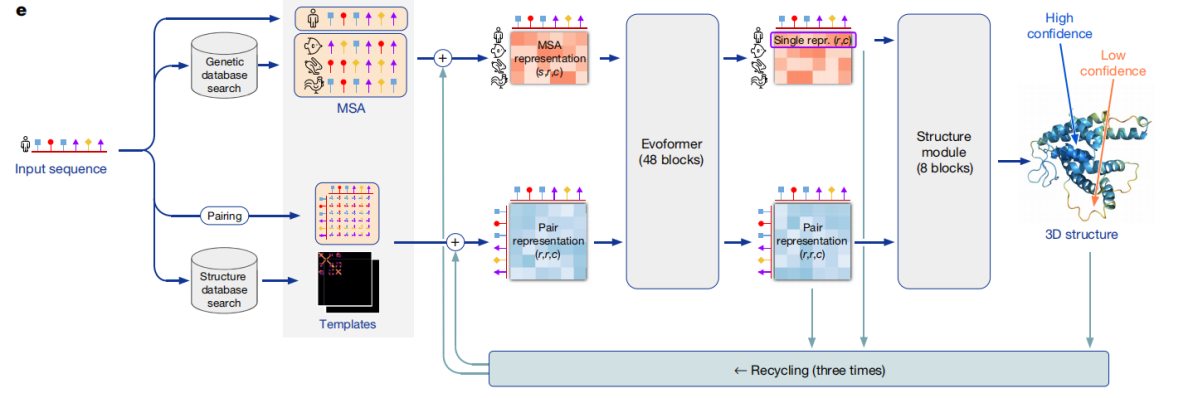
5.26 MB
imgs/header.jpg
0 → 100644
{kind=link}

145 KB
monomer.fasta
0 → 100644
multimer.fasta
0 → 100644
notebooks/AlphaFold.ipynb
0 → 100644
readme_imgs/alphafold2.png
0 → 100644
{kind=link}
130 KB
readme_imgs/alphafold2_1.png
0 → 100644
{kind=link}
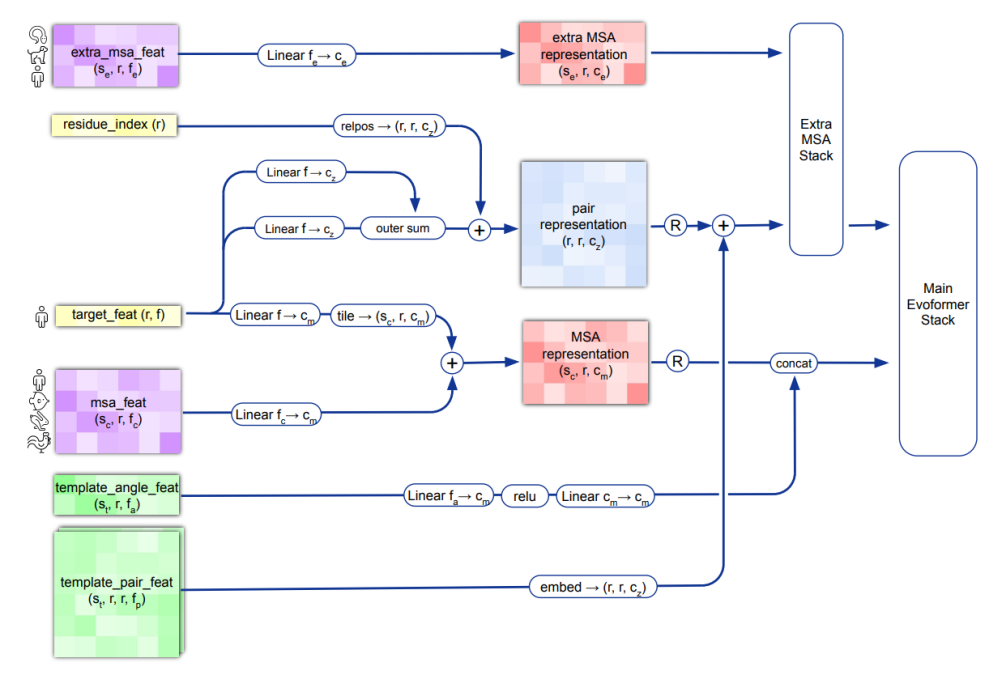
134 KB
requirements.txt
0 → 100644
| absl-py==1.0.0 | ||
| biopython==1.79 | ||
| chex==0.1.86 | ||
| dm-haiku==0.0.12 | ||
| dm-tree==0.1.8 | ||
| docker==5.0.0 | ||
| immutabledict==2.0.0 | ||
| jax==0.4.26 | ||
| ml-collections==0.1.0 | ||
| numpy==1.24.3 | ||
| pandas==2.0.3 | ||
| scipy==1.11.1 | ||
| tensorflow-cpu==2.16.1 |
requirements_dcu.txt
0 → 100644
run_alphafold.py
0 → 100644
run_alphafold_test.py
0 → 100644
run_monomer.sh
0 → 100644
run_multimer.sh
0 → 100644
scripts/download_all_data.sh
0 → 100755
scripts/download_bfd.sh
0 → 100755